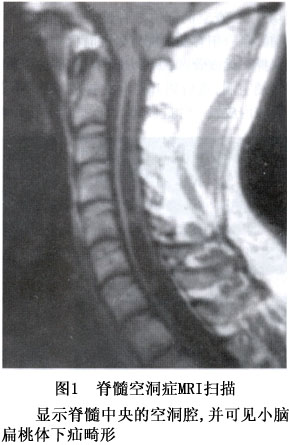
发病机制
发病机制
发病机制:
1.先天性病因 大体有四种学说:
(1)Gardner的流体力学理论:1958年开始,Gardner报道了大量Chiari Ⅰ型畸形伴脊髓空洞症的病例,他推测由于枕大孔区的梗阻(先天畸形或蛛网膜炎等),使脑脊液不能从四脑室流出,脑脊液在脉络膜丛动脉源性搏动的作用下,就会不断冲击脊髓中央管,使其扩大,并破坏中央管周围的灰质,形成空洞。手术中也发现四脑室与中央管是交通的;部分病人脑室造影时,可观察到造影剂经四脑室进入中央管;经皮肤穿刺向空洞内注入空气,气体也可溢入四脑室;而且空洞内液体蛋白含量低,与脑脊液相似。但也有不同意见:部分病人在造影、手术及尸检中未发现四脑室与中央管的交通;腰穿注入造影剂虽不流入四脑室,但空洞可显影;部分空洞与中央管分离,并且是多房的。有的学者计算了脉络丛动脉源性搏动的压力,发现压力很小,不可能造成空洞。另外此学说无法解释延髓空洞症的产生。
(2)Williams的颅内与椎管内压力分离学说:1969年以来,Williams进行了一系列研究,对脑室、空洞及蛛网膜下腔做了压力测定。认为人在咳嗽,打喷嚏及用力时,可引起颅内及椎管内静脉压上升,使脑脊髓蛛网膜下腔的压力随之升高,此时正常人是通过脑脊液在蛛网膜下腔的往返流动来平衡的。而有小脑扁桃体轻度下疝的病人,由于脑脊液循环障碍,就出现了压力的不平衡。Williams发现在咳嗽初期,腰部蛛网膜下腔的压力高于基底池,以后则相反。由此他推测小脑扁桃体可能有活瓣作用,当脊髓蛛网膜下腔压力升高时,脑脊液可上推下疝的扁桃体而流入颅内;随着脊髓蛛网膜下腔压力的下降,小脑扁桃体再度下疝,使脑脊液不能回流,造成
颅内压增高,促使脑脊液从四脑室向中央管灌注,这是颅内与脊髓、中央管与脊髓外产生的压力差,Williams称为脑脊液压力分离。这个压力差反复间断地作用很多年,就可以形成交通性脊髓空洞症。通过空洞穿刺及动物实验也发现空洞内压力高的特点。部分病人在咳嗽用力时,也有症状加重的临床报道。脊髓空洞症病人中央管与空洞的交通不是一直开放的,由于枕大孔处组织反复受压及其他原因,可以关闭。由此在空洞的进展上提出了脑脊液冲击理论,在枕大孔明显受压的病人,当咳嗽等用力时,脊髓蛛网膜下腔的压力突然升高,由于不能向颅内传递,就向脊髓内空洞传递,因空洞的开口被关闭或有活瓣作用,空洞内液体不能流入颅内,就向上面中央管旁的灰质冲击,久而久之,空洞逐渐向上扩展,并在脊髓空洞的基础上形成延髓空洞。由此说明延髓空洞不可能单独存在,此点与临床观察相符。
(3)脑脊液脊髓实质的渗透学说:1972年Ball在脊髓空洞症的尸检中发现脊髓实质内血管周围间隙的明显增宽,他在空洞内注入墨汁可沿血管周围间隙扩散,并在局部形成一些小池,特别是脊髓背侧白质明显。由此推测由于枕大孔区畸形,静脉压及脊髓蛛网膜下腔压力反复一过性的升高,长期作用于脊髓,使血管周间隙逐渐扩大,脑脊液由此渗入而形成空洞。1979年Aboulker提出神经轴突组织是能透过水的,脑脊液可沿神经组织向脊髓内渗透。临床上也有报道术中确实证实四脑室与中央管无交通的病人,在延迟脑脊液造影像上空洞可显影,而且部分空洞远离中央管,多偏在靠近脊髓表面最近的后角部分等。
(4)循环障碍学说:Netsky在脊髓空洞症病人尸检中发现髓内有血管异常,特别是后角明显。他推测随着年龄的增长,异常血管周围可发生循环障碍而导致空洞。脊髓对脑脊液的灌注或冲击的损害有保护性机制,胶质纤维增生,这些纤维会影响脊髓实质的血液供应,缺血可能是空洞产生及进展的原因之一。脊髓实质(主要在后角)先天性异常并不是发病的惟一因素。脊髓后角先天异常,加之枕大孔区畸形及静脉压等因素,使脑脊液很容易从先天异常侧的脊髓后根侵入,在局部形成空洞,随着空洞的扩展可与中央管交通,继之中央管逐渐扩大,最后可与四脑室相通。
2.后天性病因 多由脊髓肿瘤、蛛网膜炎及外伤等因素引起。外伤可使脊髓中心部坏死,造成渗出液及破坏产物的积聚,使渗透压升高,液体潴留,由于髓内压力升高,可破坏周围组织,使空洞逐渐扩大。动物实验中发现,在切断的脊髓断端附近出现一些微小囊肿,由此可推测这些囊肿的破裂、汇合可能是空洞形成的原因。对于蛛网膜炎后的脊髓空洞症,主要由于缺血及静脉栓塞造成。脊髓肿瘤引起的脊髓空洞症主要与肿瘤细胞分泌蛋白性液体有关。
脊髓空洞症发病机制复杂,枕大孔区畸形或梗阻是导致空洞形成的重要因素之一。由于每个人的病因、体质及机体代偿能力的不同,空洞的形成和发展也各不相同,所以应根据临床特点及病期的不同,进行不同病因探讨及综合分析。
脊髓空洞症多发生于颈段及上胸段的中央管附近,靠近一侧后角,形成管状空洞,可延续多个脊髓节段,并不一定与中央管相通。在脊髓横断面上,可见空洞腔占据了大部分的髓质,前角背侧也可受累,前后连合结构常被破坏。随着空洞腔的进一步发展,后角也可受累、甚至包括后索的腹侧。空洞可局限于脊髓的一侧,也可占据两侧。空洞形状不一,在脊髓同一平面有可能存在多个空洞腔,它们可相互隔开,也可互相联通。此症有的与延髓空洞同时存在。空洞向上有延至脑桥与中脑者。腰段以下空洞症较少见。少数情况于脊髓末端见有小的空洞,并且与
脊柱裂共存。
脊髓受压变性常是空洞扩大之必然结果。空洞部位脊髓呈梭形膨隆,颜色变淡,软膜血管减少。空洞可位于中央或偏于一侧,或偏于前或后,使脊髓灰质、侧索、后索受压变性。空洞之壁光滑,为增生的胶质及趋于变性的神经纤维,颜色变白,周围的神经纤维呈现水肿。晚期脊髓空洞巨大者,脊髓组织菲薄,可造成椎管腔的梗阻。
依照病理状况,脊髓空洞症可分为两种类型:一类为交通性脊髓空洞症,即脊髓空洞与第四脑室、蛛网膜下腔脑脊液相交通,常合并小脑扁桃体下疝Ⅰ型与Ⅱ型畸形。它可能系生长发育过程中的某些异常因素的作用所致,例如脊髓中央管可能在较高的脑脊液压力的作用下,液体不断渗漏入周围神经组织,使之发生持续性扩张而形成本病;另一类为非交通性脊髓空洞症,空洞与脑脊液循环通路不相交通。它的形成与髓内肿瘤、外伤性截瘫和一些变性疾病有一定关系。